

Retrosynthesis analysis is a critical task in organic chemistry and central to many important industries. It primarily involves decomposing a target molecule into commercially available molecules step by step. Since synthesis strategies can be quite diverse and strategic, retrosynthesis planning with expert knowledge has long been considered an “art.”
Recently, machine learning-based approaches have achieved promising results on this task, particularly in single-step retrosynthesis prediction. In retrosynthesis, a molecule can be represented as either a 2D graph or a 1D SMILES (simplified molecular-input line-entry system) sequence. SMILES is a notation system used to represent chemical structures using plain text, which consists of a sequence of characters to describe the arrangement of atoms, bonds, and rings within a molecule. SMILES can be considered a traversal on the corresponding molecular graph, as shown in Figure 1.
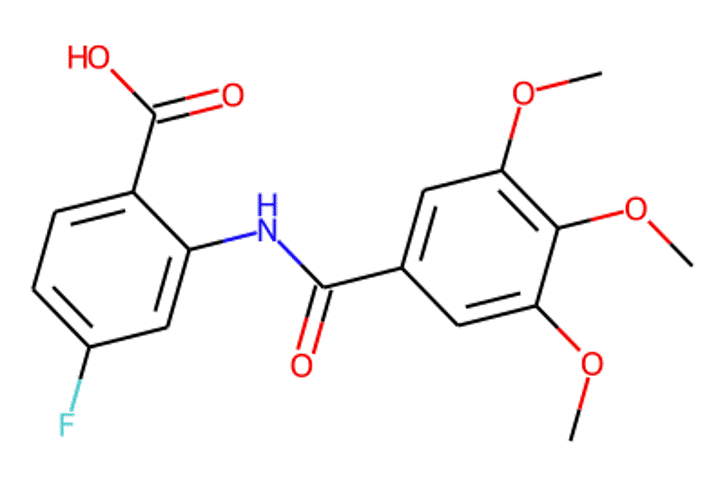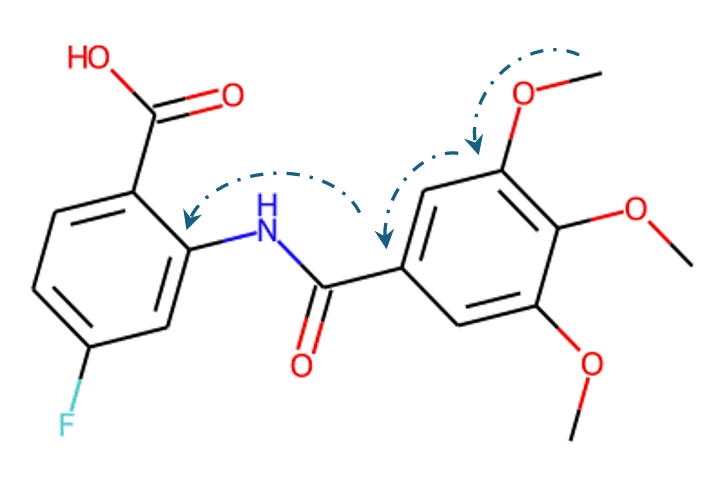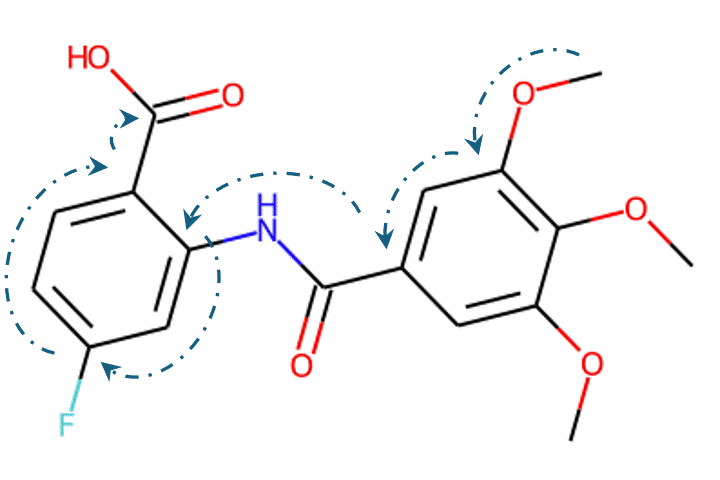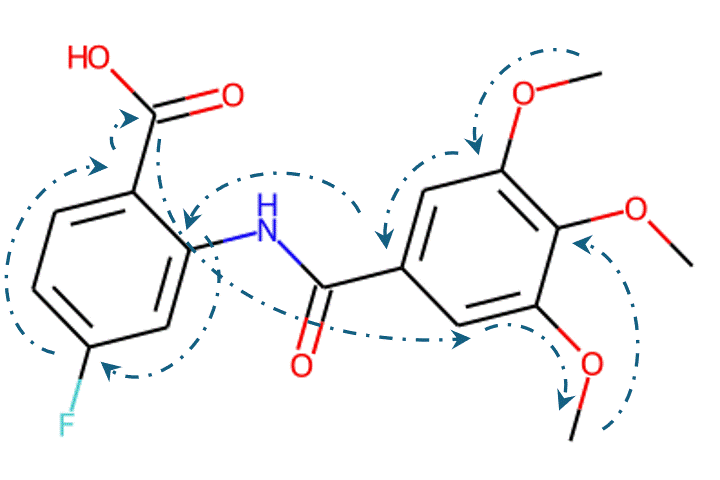
Given the representations of molecules, most machine learning-based approaches employ encoder-decoder frameworks, where the encoder part encodes the molecular (the target product) sequence or graph as high dimensional vectors, and the decoder takes the output from the encoder and generates the output sequence (the predicted reactant) token-by-token autoregressively.
Casting retrosynthesis analysis as a sequence decoding problem enables the use of deep neural architectures that are well-developed in machine translation or graph neural networks. While AI has made significant strides in predicting reactants, it’s crucial to acknowledge the expertise of human chemists. In real-world route scouting tasks, synthetic chemists rely on their professional experience and abstract understanding of underlying mechanisms. They often start with molecular substructures or fragments that are chemically similar to target molecules, providing clues for a series of chemical reactions that may yield the target product.
Our paper, Single-step retrosynthesis prediction by leveraging commonly preserved substructures (opens in new tab), proposes a novel approach that leverages commonly preserved substructures in organic synthesis. This approach incorporates chemists’ insight in retrosynthesis, bringing the AI model closer to the way human experts think.
Substructure extraction and modeling
In the context of organic chemistry, “substructures” refer to molecular fragments or smaller building blocks that are chemically similar or preserved within target molecules. These substructures serve as essential components for understanding the assembly of complex molecules and play a significant role in retrosynthesis analysis.
Based on this concept, our framework consists of three main modules:
- Reaction Retrieval: This module retrieves similar reactions, given a product molecule as a query. It uses a learnable cross-lingual memory retriever to align reactants and products in high-dimensional vector space.
- Substructure Extraction: We extract the common substructures from the product molecule and the top cross-aligned candidates, based on molecular fingerprints. These substructures provide a reaction-level, fragment-to-fragment mapping between reactants and products.
- Substructure-level Sequence-to-Sequence Learning: We convert the original token-level sequence to a substructure-level sequence. The new input sequence includes the SMILES strings of the substructures followed by the SMILES strings of other fragments with virtual number labels. The output sequences are the fragments with virtual numbers. The virtual numbers are used to indicate the bond breaking/connecting site.

Unlike most existing work, our model only needs to predict the fragments connected to the substructure, thereby simplifying the prediction task, with the substructure part remaining unchanged.
In the example shown in Figure 2, the substructure “COC(=O)Cc1cc2ccc(F)cc2[2cH]c1C.C[1SH](=O)=O” remains unchanged, and the model only needs to predict that the fragment “[2BH]2OC(C)(C)C(C)(C)O2.[1cH]1ccc(Br)nc1”. The substructure SMILES and the predicted fragment SMILES are then combined to form a complete reactants SMILES.
Spotlight: Event Series

Microsoft Research Forum
Join us for a continuous exchange of ideas about research in the era of general AI. Watch the first four episodes on demand.
Retrosynthesis prediction
We analyzed our method using the USPTO full dataset (opens in new tab) and compared it to other notable works in the field. In almost every scenario, our method achieved comparable or better top-1 accuracy compared to previously tested methods. On the subset of data where substructures were successfully extracted, model performance significantly improved compared to the overall result.
The improvement in our method can be attributed to two main factors:
- Our method managed to successfully extract substructures from 82.2% of all products on the USPTO full test dataset, demonstrating the general applicability of this approach.
- We only needed to generate fragments connected to virtually labeled atoms in the substructures, which shortened the string representations of molecules and significantly lowered the number of atoms to be predicted.

A key aspect of our method for one-step retrosynthesis is the extraction of product-specific substructures. By doing so, we can better capture subtle structural changes from reactants to products that are unique to each reaction. Take phthalimide, a common heterocyclic substructure, as an example. We analyzed four exemplary reactions where the reactants contain phthalimide (see Figure 3). The extracted substructures vary among different reaction types, demonstrating the product-specific nature of the substructures.
In reaction (a) and reaction (b), phthalimide is not considered part of the substructure because it incorporates the reaction. However, in reaction (c) and reaction (d), the substructures are different, yet they both contain phthalimide. These results show that substructures are indeed product-specific, which aligns with our expectations.
Incorporating human insights into decision-making
In addition, leveraging commonly preserved substructures offers another benefit: providing users with valuable insights for decision-making in retrosynthesis planning. When compared to existing methods, our approach can help human experts assess potential pathways and eliminate infeasible reactions using their chemistry knowledge.
For each input product molecule, we extract multiple substructures from retrieved reactions, (see details in our paper) and for some cases, not all substructures are correct. As such, we can group predictions by substructures. As shown in Figure 4, the predicted groups of reactants and reactions offer valuable information to experts. For instance, they can refine predictions by comparing reactions associated with retrieved candidates, making our predictions more explainable and trustworthy compared to existing “black-box” models.

We hope that our work will spark interest in this fast-growing and highly interdisciplinary area of retrosynthesis prediction and other related topics. By pushing the boundaries of what’s possible in chemistry and machine learning, we can continue to make strides in understanding complex chemical reactions and designing more efficient retrosynthetic strategies.



